










字节跳动研发并开源了基于gpu加速的大规模量子化学计算工具集byteqc,显著提升了量子化学计算效率。该工具集在现代gpu上高效实现了多种标准量子化学算法,并结合量子嵌入方法,在保持高精度的前提下,大幅扩展了可计算体系的规模。

- 论文链接:https://www.php.cn/link/f0a94cc746c82ccf258040d541a2e15e
- 代码链接:https://www.php.cn/link/f0a94cc746c82ccf258040d541a2e15e
ByteQC的核心优势在于其高效的GPU实现和创新的算法策略。通过引入NVIDIA的cuTENSR/cuTENSORMG张量计算库,并优化缓存及原位操作,ByteQC最大限度地减少了显存占用,并提升了计算速度。针对GPU架构特点,ByteQC团队还开发了基于warp特例化的动态生产者-消费者模型和基于warp同步原语的竞态求和算法,有效解决了复杂计算逻辑的GPU实现难题。
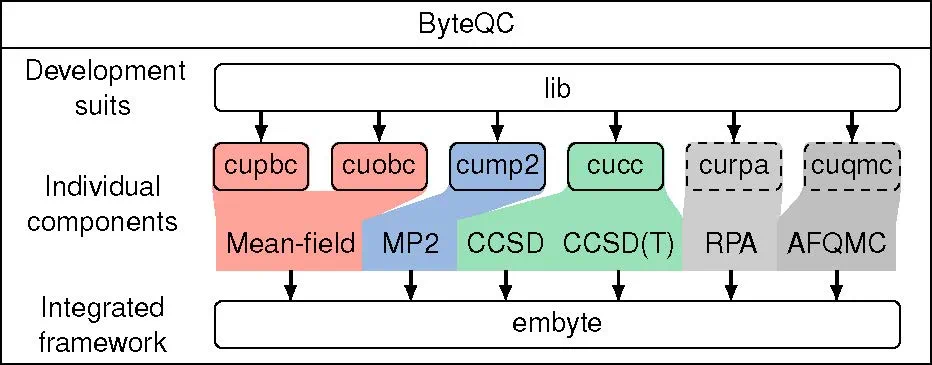 图1. ByteQC软件架构
图1. ByteQC软件架构
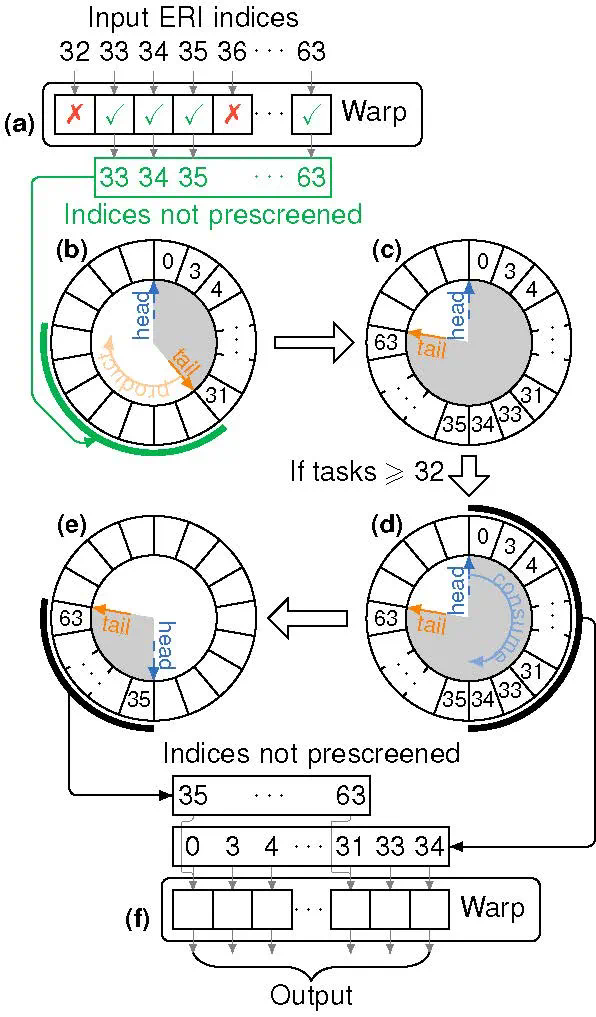 图2. 基于warp特例化的生产者-消费者模型
图2. 基于warp特例化的生产者-消费者模型
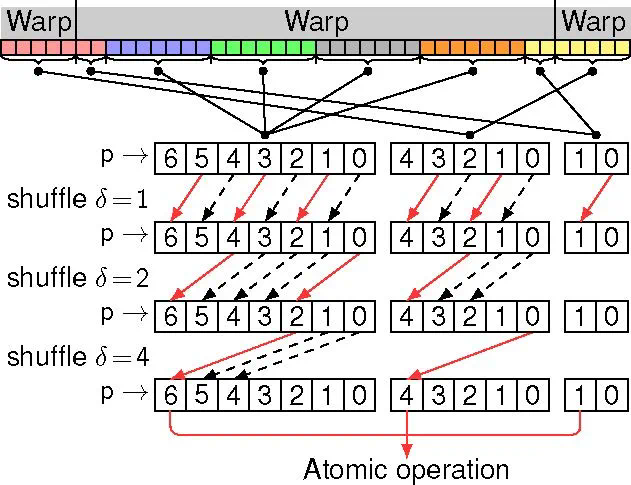 图3. 基于warp同步原语的相邻7个线程的竞态求和
图3. 基于warp同步原语的相邻7个线程的竞态求和
基准测试结果显示,ByteQC在单A100 GPU上,标准量子化学算法最高可实现60倍于100核CPU的加速,且多卡扩展性良好。可计算体系规模也显著提升,例如CCSD(T)计算可达1380轨道,MP2计算可达11040轨道,平均场计算甚至可超过10万轨道。
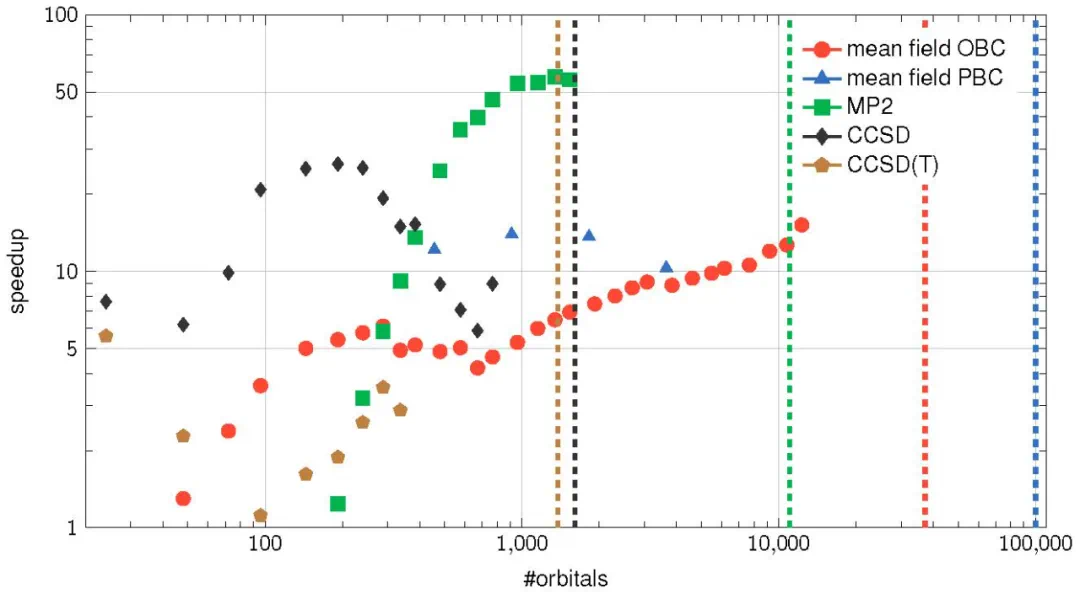 图4. ByteQC的子模块加速比(数据点)和计算规模(虚线)
图4. ByteQC的子模块加速比(数据点)和计算规模(虚线)
结合量子嵌入方法,ByteQC成功实现了对2753轨道水团簇和3929轨道氮化硼表面水吸附体系的CCSD(T)高精度计算,验证了其在真实材料计算中的应用潜力。
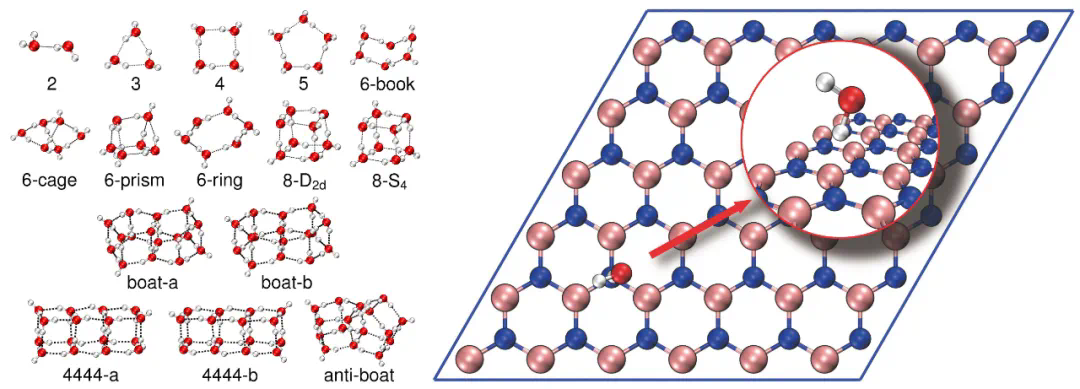 图5. (左)水团簇结构和(右)氮化硼表面水吸附结构
图5. (左)水团簇结构和(右)氮化硼表面水吸附结构
ByteQC的开源发布,为量子化学计算领域提供了强有力的工具,有望加速相关领域的研究进展。